Text-mining to identify key molecular targets associated with diabetes.
NLP
Disease-gene association
Python
Author
Manish Datt
Published
September 5, 2024
Diabetes is a synergistic manifestation of a multitude of metabolic dysfunctions. This makes it difficult to pinpoint a specific molecular target for therapeutic interventions. Over the years, several drugs have been approved targeting different proteins. The best way to get information about the major proteins involved in the pathophysiology of Diabetes is to review the scientific literature published in this field. Here, we’ll mine the research articles in PubMed to identify major proteins for which inhibitors have been (or are being) developed to treat diabetes. Natural Language Processing (NLP) models trained on a corpus of biomedical data will be used to parse the literature.
Searching PubMed
We’ll search for all the research articles with “diabetes” in the title and “inhibitor” (and its variants) in the title or abstract. The functions available in the Biopython library will be used to do the search. Detailed steps for searching PubMed are outlined in this post. The full PubMed search query is given below.
((((diabetes[Title]) AND (inhibit[Title/Abstract] OR inhibitor[Title/Abstract] OR inhibitors[Title/Abstract])) NOT (review[Publication Type])) NOT (Clinical Trial[Publication Type])) NOT (Systematic Review[Publication Type])
Show Python code
from Bio import EntrezEntrez.email ='example@test.com'#change email idpubs_diab = Entrez.esearch(db="pubmed", term="((((diabetes[Title]) AND (inhibit[Title/Abstract] OR inhibitor[Title/Abstract] OR inhibitors[Title/Abstract])) NOT (review[Publication Type])) NOT (Clinical Trial[Publication Type])) NOT (Systematic Review[Publication Type])",\ retmode="xml", retmax=9999) record = Entrez.read(pubs_diab)pubs_diab.close()idlist = record["IdList"]withopen('pubmed_ids.txt', 'w') as f:for ids in idlist: f.write(ids +'\n')# get the abstractswithopen('pubmed_ids.txt', 'r') as f: idlist = [line.strip() for line in f]def fetch_abstracts(pmid): abstracts = [] pub = Entrez.efetch(db="pubmed", id=pmid, rettype="medline", retmode="text") record = pub.read() pub.close() lines = record.splitlines()if'AB - 'in record:for i, line inenumerate(lines):if line.startswith('AB - '): abstract = line[6:].strip() j = i +1while j <len(lines) and lines[j].startswith((' ', '\t')): abstract +=' '+ lines[j].strip() j +=1 abstracts.append(abstract)else: abstracts.append('No abstract available')return abstractsresults = []for ids in idlist: res = fetch_abstracts(ids) results.extend(res[0].splitlines())# save the abstracts to a filewithopen('abstracts_diabetes.txt', 'w',encoding='utf-8') as f:for abstract in results: f.write(abstract +'\n')
As of 31 August 2024, this search returned 9,966 hits; and 9,335 entries had abstracts in PubMed. Using these abstracts we’ll now perform the NLP analysis.
NLP based Named-Entity Recognition
Natural Language Processing falls under the umbrella of AI and aims to enable computers to understand human languages. There are several techniques that contribute to an efficient NLP analysis. One of these is Named-Entity Recognition (NER) wherein a pre-trained model detects and labels entities to different words in a sentence. For example, the table below shows the NER for the following sentence.
India’s largest listed biopharmaceutical firm Biocon Ltd forged a strategic deal with Pfizer Inc. for worldwide commercialization of four insulin products, seeking to address a market worth a combined $14 billion.
Entity
Label
Explanation
India
GPE
Geopolitical entities, including countries, cities, and states.
Biocon Ltd
ORG
Organizations, including companies, agencies, and institutions.
Pfizer Inc.
ORG
Organizations, including companies, agencies, and institutions.
four
CARDINAL
Cardinal numbers, such as “one,” “two,” etc.
$14 billion
MONEY
Monetary values, including amounts and currencies.
The above NER was done using en_core_web_sm which is a pre-trained model on English text. Custom models can be trained on a domain-specific text as well. For instance, models can be trained with biomedical text to identify entities such as genes, diseases, species, etc. There are now quite a few pre-trained NLP models for biomedical text, such as BioBERT, BioGPT, SciSpacy, etc. These models along with an NLP framework (like spacy) make it easy to set up a biomedical text-mining workflow.
The example below shows the named-entities identified in two sentences using en_ner_bionlp13cg_md model available in spacy.
With regards to medication management, for patients with clinical cardiovascular disease, a sodium-glucose cotransporter 2 (SGLT2) inhibitor or a glucagon-like peptide 1 (GLP-1) receptor agonist with proven cardiovascular benefit is recommended.
Entity
Label
patients
ORGANISM
cardiovascular
ANATOMICAL_SYSTEM
sodium-glucose cotransporter 2
GENE_OR_GENE_PRODUCT
SGLT2
GENE_OR_GENE_PRODUCT
glucagon-like peptide 1
GENE_OR_GENE_PRODUCT
We also quantified PAGs before and after glucose control with a sodium-glucose cotransporter 2 (SGLT2) inhibitor, dapagliflozin.
Entity
Label
glucose
SIMPLE_CHEMICAL
sodium-glucose cotransporter 2
GENE_OR_GENE_PRODUCT
SGLT2
GENE_OR_GENE_PRODUCT
dapagliflozin
SIMPLE_CHEMICAL
Next, we’ll run this code on all the abstracts collected above to identify GENE_OR_GENE_PRODUCT in those followed by counting the frequency of different genes. Below are the top five genes with maximum number of counts.
Show Python code
import spacyfrom collections import Counternlp = spacy.load("en_ner_bionlp13cg_md")withopen('abstracts.txt', 'r',encoding='utf-8') as f: abstracts = [line.strip() for line in f]abstracts = [x for x in abstracts if x !="No abstract available"]#print("Number of abstracts:",len(abstracts))all_genes = []for abs1 in abstracts: doc = nlp(abs1) genes_list = [ent.text for ent in doc.ents if"GENE"in ent.label_] all_genes.extend(set(genes_list))# save the gene counts to a filewithopen('gene_counts.txt', 'w',encoding='utf-8') as f:for gene, count in Counter(all_genes).items(): f.write(f"{gene}: {count}\n")# display top 5df = pd.DataFrame.from_dict(Counter(all_genes), orient="index")df = df.reset_index()df.columns=["Gene", "Count"]df = df.sort_values('Count', ascending=False, ignore_index=True)display(df.head())
Gene
Count
insulin
2525
SGLT2
790
hemoglobin
621
T2D
560
DPP-4
435
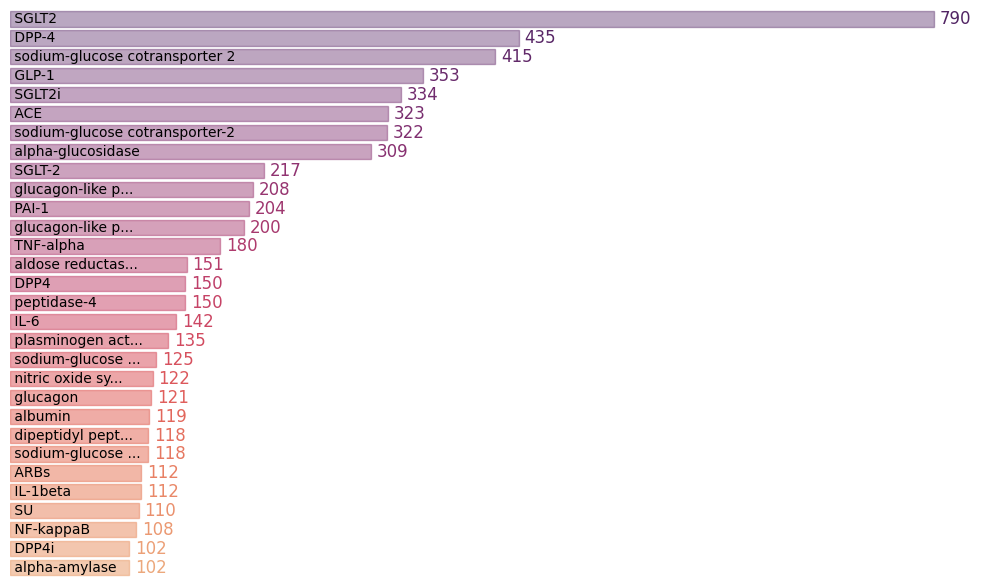
As you would have noticed, there are genes in this list that are irrelevant to our analysis ー entries like insulin, hemoglobin, T2D. Insulin is, for sure, associated with diabetes but can’t be a drug target; the same applies to hemoglobin. So, we need to filter this list to exclude such entries. After manually editing the list, the frequencies for different genes with counts >100 are shown below.
Show Python code
import pandas as pdimport matplotlib.pyplot as pltimport seaborn as snsimport numpy as npdata = []withopen('gene_counts - copy.txt', 'r',encoding='utf-8') as f:for line in f:if line.count(':') ==1: gene, count = line.strip().split(':') data.append((gene.strip(), int(count.strip())))# create a DataFramedf = pd.DataFrame(data, columns=['Gene', 'Count'])# add counts where gene name is the same. keep the first rowdf = df.groupby('Gene').agg({'Count': 'sum'}).reset_index()df = df.sort_values('Count', ascending=False, ignore_index=True)exclude_list = ["insulin", "hemoglobin", "t2d"]df = df[~df['Gene'].str.lower().isin(exclude_list)]df['percent'] = (df['Count'] /9335) *100df_subset = df[df['Count'] >100]plt.figure(figsize=(10, 6))colors = sns.color_palette('flare_r', len(df_subset))a_values = np.linspace(0.4, 0.6, len(df_subset)).tolist()sns.barplot(x='Count', y='Gene', data=df_subset)for i, bar inenumerate(plt.gca().patches): bar.set_color(colors[i]) bar.set_alpha(a_values[i])all_counts = {}for i, v inenumerate(df_subset['Count']): plt.text(v +5, i, str(v), color=colors[i], ha='left', va='center', fontsize=12) #(df['percent'].values[i])[:4] all_counts[i] = vfor i, v inenumerate(df_subset['Gene']):iflen(v) >15and all_counts[i]<300: v = v[:15] +'...' color_text = sns.color_palette('gray_r', len(df_subset))[i] plt.text(0, i, " "+v, color="black", ha='left', va='center')plt.axis('off')plt.box(False)plt.tight_layout()plt.show()
There are still some outstanding issues with these gene frequencies. As you can see, there are separate bars for different gene synonyms. For instance, SGLT-2, sodium-glucose cotransporter 2, and sodium-glucose cotransporter-2 have separate counts. Subtle differences in the gene name (like the presence or absence of a dash) result in their segregation. The table below highlights the synonymous entries for SGLT-2 in yellow, and GLP-1 in pink; not to mention several other “typo” variants in this list.
Gene
Count
Gene
Count
SGLT2
790
peptidase-4
150
DPP-4
435
IL-6
142
sodium-glucose cotransporter 2
415
plasminogen activator inhibitor-1
135
GLP-1
353
sodium-glucose co-transporter 2
125
SGLT2i
334
nitric oxide synthase
122
ACE
323
glucagon
121
sodium-glucose cotransporter-2
322
albumin
119
alpha-glucosidase
309
dipeptidyl peptidase 4
118
SGLT-2
217
sodium-glucose co-transporter-2
118
glucagon-like peptide-1 receptor agonists
208
ARBs
112
PAI-1
204
IL-1beta
112
glucagon-like peptide-1
200
SU
110
TNF-alpha
180
NF-kappaB
108
aldose reductase
151
DPP4i
102
DPP4
150
alpha-amylase
102
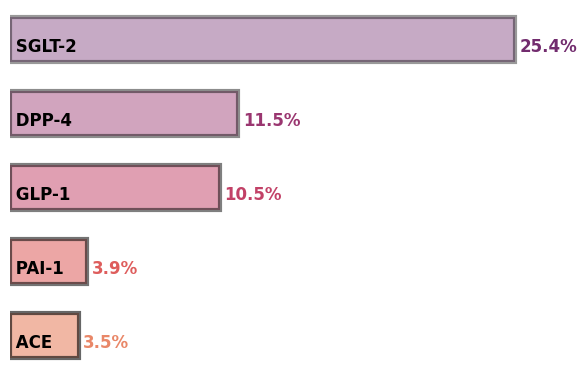
Now, we need to manually curate this list by renaming the synonymous genes to a particular name so that we can add their counts. After all the hard work, below are the top five genes identified using text-mining.
NLP opens up exciting opportunities to programmatically parse the ever-expanding repertoire of biomedical text. Meticulous use of NLP techniques can indeed provide new insights based on knowledge stored in literature databases. There are, however, some caveats that one needs to keep in mind when working with such AI capabilities. First and foremost is the model training. The named-entity recognition in biomedical text is still limited to a few entities. This limits the kind of questions that can be addressed using these models. Also, some words like T2D are labeled as a gene when they should be labeled as a disease. Another issue is the varied nomenclature for a particular thing in biomedical text. For example, GLP1 and GLP-1 are identified as two different genes. In fact, this point actually holds a lesson for biologists! They need to be mindful with the usage of technical terms in manuscripts and should adhere to canonical names to avoid littering the databases with undesired aliases.
This post walks through the method of NLP-based Text and Data Mining (TDM) and has flagged important issues along the way. The dataset used here is rather small with ~10K abstracts. Richer data, particularly full-text articles, would certainly add depth to the analysis. With the enhancement of the NLP models and the availability of open-access literature, TDM is poised to propel biomedical discoveries in the future.